SMARCA4
Research question
This task-based tutorial builds upon three previous tool-oriented tutorials build_bedpe, query_bedpe, and plot_contacts and also introduces an alternate solution using get_loops. Becoming familiar with these tools is recommended before diving into this tutorial. In this tutorial, we will explore two different approaches to answer the following questions:
How do the genomic interactions between H3K4me3 and H3K27ac peaks associated with the SMARCA4 gene differ between a control sample and a case sample?
To answer this question, we will use mm10 genes sourced from UCSC, coordinates for the SMARCA4 gene, and pre-existing peak files for H3K27ac and H3K4me3. First, we will use bedtools to extract the H3K4me3 peaks that are located in the gene body. Next, we will use build_bedpe to create genomic pairings between those peaks and all H3K27ac peaks within a user-defined genomic range. We’ll use query_bedpe to obtain the contact frequencies of the pairings and plot_contacts to visualize our process. We will conclude this tutorial with visualizations of the strongest and weakest interactions between control and case samples.
To list all available sample data on the Tinkerbox, simply type the following command into the terminal window:
list_samples.shLet’s assign variable names to our samples and reference files for ease
control=WT_K27ac_2i_Rep1
case=A2_K27ac_2i_Rep1It might also be handy to have a list of mm10 genes, that’ll allow us to ask meaningful biological questions. We obtained mm10 genes from UCSC
gene_list=~/shared-files/UCSC.genes.mm10.bedObtain SMARCA4 gene coordinates
Since we now have a gene file handy, we can obtain the SMARCA4 gene coordinates from our gene list
gene=Smarca4
cat $gene_list | grep $gene > ${gene}.bed
cat ${gene}.bedObtain reference files from ENCODE-3
We also have some pre-cooked reference files ready; mESC H3K27ac and H3K4me3 peaks from ENCODE-3-
K4me3=~/path/to/E14Tg2a_K4me3_ENCSR212KGS_5kbins_merged_mm10.bed
K27ac=~/path/to/C57BL6_K27ac_ENCSR775OEV_5kbins_merged_mm10.bedExtract H3K4me3 peaks
As we now have the SMARCA4 gene body, we can extract the H3K4me3 peaks that are located in the gene body, using bedtools
bedtools intersect \
-a ${gene}.bed \
-b $K4me3 \
-wa -wb \
| cut -f 5-7 > ${gene}_K4me3.bedConstruct pairs
Using build_bedpe, we can now construct genomic pairings (as a bedpe file) between our SMARCA4 K4me3 peak of interest and all H3K27ac peaks within a certain genomic distance.
build_bedpe.sh \
-A ${gene}_K4me3.bed \
-B $K27ac \
--min_dist 30000 \
--max_dist 300000 > ${gene}_K4me3-K27ac_pairings.bedpe
cat ${gene}_K4me3-K27ac_pairings.bedpe
bedpe=${gene}_K4me3-K27ac_pairings.bedpeObtain contact frequencies
Great! We now have all the genomic pairings we want! We can now obtain the contact frequencies of these pairings using query_bedpe and sort them in increasing order, based on control vs case values.
query_bedpe.sh \
--sample1 $control
--sample2 $case \
--pair $bedpe \
--formula sum \
--genome mm10 \
--norm cpm \
--resolution 5000 \
| sort -k9n > ${gene}_K4me3-K27ac_pairings_annotated.bedpe
cat ${gene}_K4me3-K27ac_pairings_annotated.bedpe
chr9 21605000 21620000 chr9 21715000 21775000 1.237 0.94 -0.297
chr9 21605000 21620000 chr9 21780000 21825000 0.989 0.715 -0.274
chr9 21605000 21620000 chr9 21665000 21710000 0.67 0.414 -0.257
chr9 21505000 21570000 chr9 21605000 21620000 1.661 1.429 -0.232
chr9 21575000 21580000 chr9 21605000 21620000 0.177 0.038 -0.139
chr9 21605000 21620000 chr9 21830000 21880000 0.389 0.301 -0.088
chr9 21365000 21375000 chr9 21605000 21620000 0.212 0.15 -0.062
chr9 21475000 21480000 chr9 21605000 21620000 0 0 0
chr9 21605000 21620000 chr9 21885000 21890000 0 0 0
chr9 21605000 21620000 chr9 21645000 21660000 0.141 0.15 0.009
chr9 21380000 21385000 chr9 21605000 21620000 0.035 0.075 0.04
chr9 21490000 21500000 chr9 21605000 21620000 0.035 0.075 0.04
chr9 21605000 21620000 chr9 21895000 21965000 0.565 0.715 0.15
chr9 21395000 21470000 chr9 21605000 21620000 0.53 0.752 0.222Visualize pairs
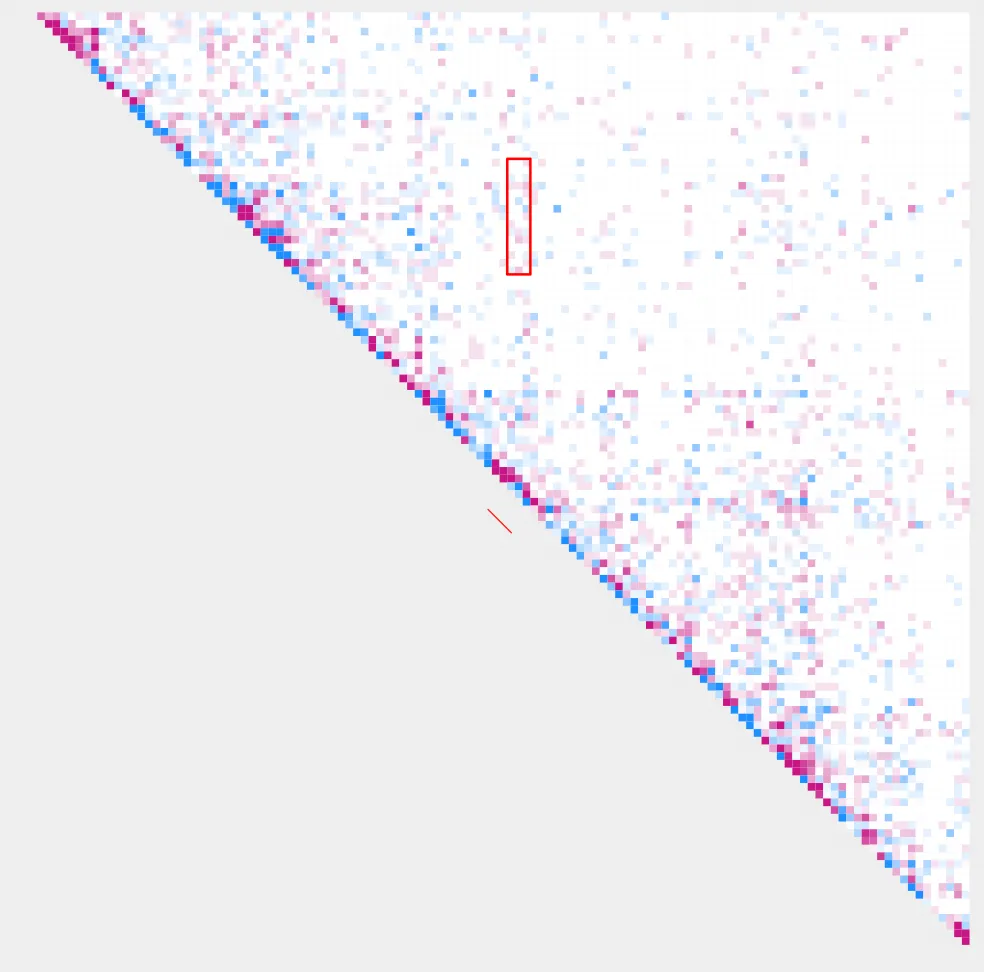
Let’s visualize our results using plot_contacts
plot_contacts.sh
--sample1 $control \
--range chr9:21300000:21900000:${gene} \
--genome mm10 \
--output_name ${gene}.pdf \
--norm cpm \
--resolution 5000 \
--max_cap 0.2 \
--bedpe $bedpe \
--bedpe_color 000000 \
--annotations_custom ${gene}_K4me3.bed \
--annotations_custom_color FF0000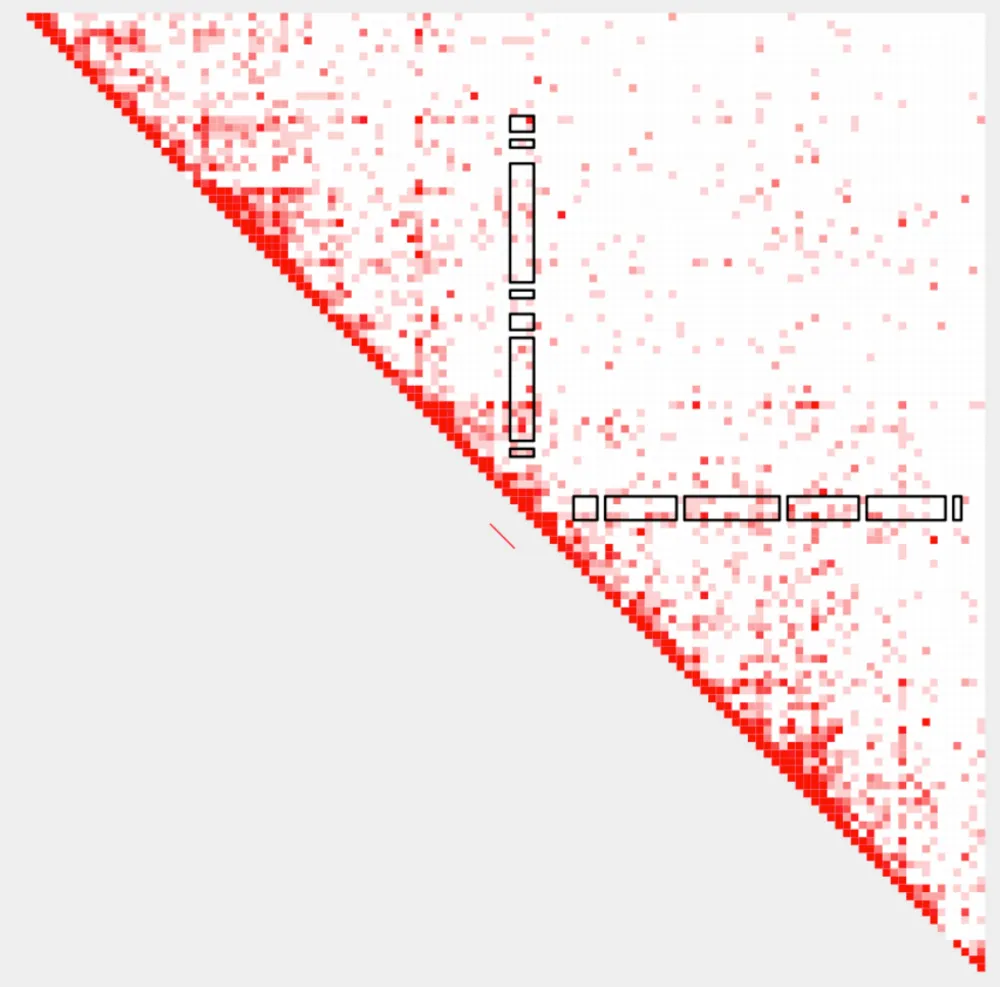
Sort pairs
Which K4me3-K27ac pairs show maximum and minimum changes in contacts across the two samples? Let’s use commands to extract the top and bottom pairings from this list. Next, we will use the --bedpe option of plot_contacts to view the high and low pairs within the contact matrix.
head -n 1 \
${gene}_K4me3-K27ac_pairings_annotated.bedpe \
> low_${gene}_K4me3-K27ac_pairings_annotated.bedpe
tail -n 1 \
${gene}_K4me3-K27ac_pairings_annotated.bedpe \
> high_${gene}_K4me3-K27ac_pairings_annotated.bedpeVisualize low contact value pairs
plot_contacts.sh \
--sample1 $control \
--sample2 $case \
--range chr9:21300000:21900000:${gene} \
--genome mm10 \
--output_name ${gene}_1.pdf \
--norm cpm \
--resolution 5000 \
--max_cap 0.2 \
--bedpe low_${gene}_K4me3-K27ac_pairings_annotated.bedpe \
--bedpe_color 0000FF \
--annotations_custom ${gene}_K4me3.bed \
--annotations_custom_color FF0000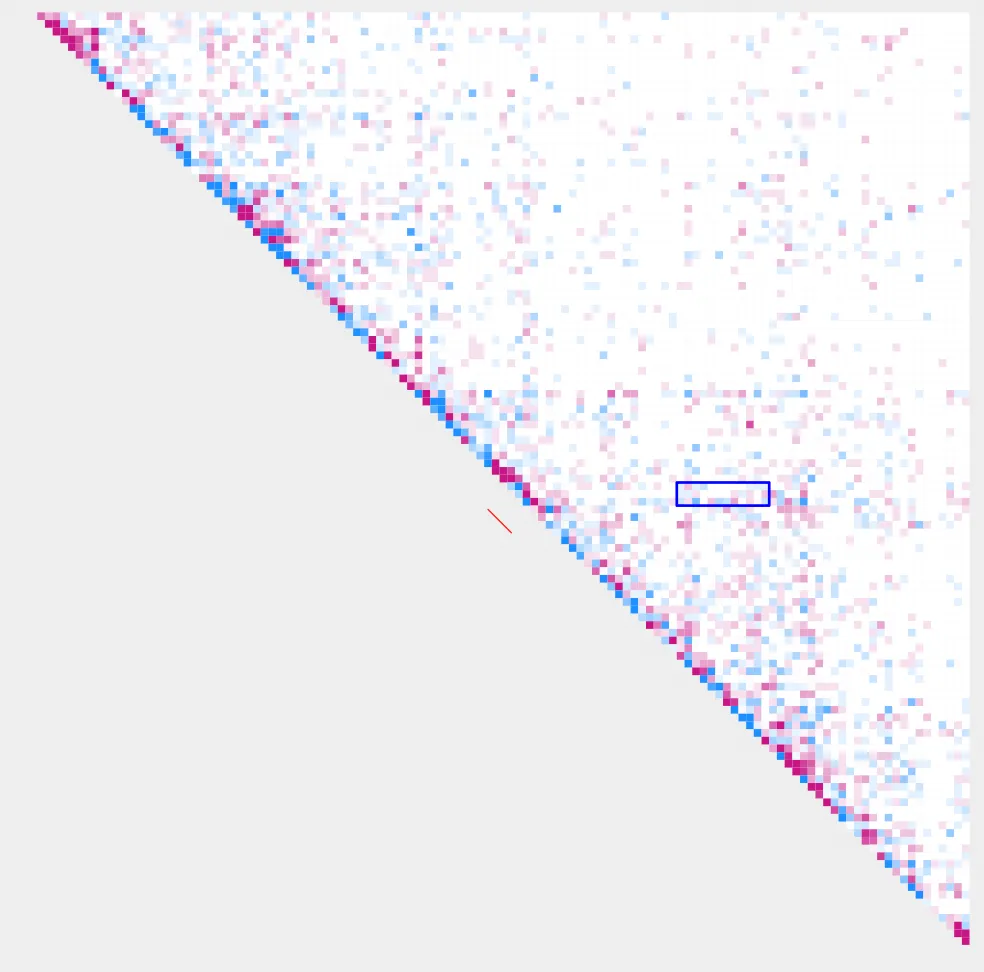
Visualize high contact value pairs
plot_contacts.sh \
--sample1 $control
--sample2 $case
--range chr9:21300000:21900000:${gene} \
--genome mm10 \
--output_name ${gene}_2.pdf \
--norm cpm \
--resolution 5000 \
--max_cap 0.2 \
--bedpe high_${gene}_K4me3-K27ac_pairings_annotated.bedpe \
--bedpe_color FF0000 \
--annotations_custom ${gene}_K4me3.bed \
--annotations_custom_color FF0000