Examples
This is a tool-oriented primer designed to guide you through the functionalities of query_bedpe. From your tinker box, type query_bedpe -h into the command window and press enter to view the help message.
Help message
query_bedpe -h
Query and annotate a bedpe file with AQuA normalized contact values
-P|--pair : Full path to the bedpe (pairs) file you want to annotate, without headers!
-A|--sample1 : Name of the sample you want to use as it appears on the Tinkerbox
-G|--genome : The genome build the sample(s) has been processed using. Strictly hg19 or hg38
[ -Q|--norm ] : Which normalization to use. Strictly 'none', 'cpm' or 'aqua' in lower case. Non-spike-in samples default to cpm. Spike-in samples default to aqua.
[ -B|--sample2 ] : The name of the second sample. If triggered, calculates the delta contact values for that pair. Useful in case vs control
[ -R|--resolution ] : Resolution of sample in basepairs, using which the contact values should be calculated. Default 5000. Accepted resolutions- 1000,5000,10000,25000,50000,100000,250000,500000,1000000,2500000
[ -f|--formula ] : Arithmetic to use to report contact calues. Options- center, max, average, sum. Default = center
[ -F|--fix ] : If FALSE, reports new coordinates based on arithmetic center or max. Default = TRUE
[ --shrink_wrap ] : Squeezes a 2D bedpe interval until supplied value is reached. Default = FALSE
[ --split ] : Splits a 2D bedpe interval into multiple sub-intervals greater than supplied value. Default = FALSE
[ --padding ] : Joins sub-intervals in 2D space reported by --split, based on supplied value in bin units. Default = 2
[ --expand ] : Expands 1D bedpe feet in both directions based on supplied value(in bin units). Default = 0
[ -I|--inherent ] : If TRUE, hic values transformed to inherent units. For one-sample tests only. Default = FALSE
[ -m|--preserve_meta]: If TRUE, bedpe metadata columns will be preserved. Default = FALSE
[ -h|--help ] : Help message. Primer can be found at [https://rb.gy/zyfjxc](https://rb.gy/zyfjxc)Viewing loops
Let’s tinker with this tool using provided sample data in your tinker box.
The sample data comes from H3K27ac HiChIP experiments performed in a childhood rhabdomyosarcoma cell line (Gryder et al., 2019), treated with DMSO and HDAC inhibitor Entinostat.
sample1=RH4_DMSO
sample2=RH4_HDACiLoops in 3D genomics are two stretches of DNA with high contact frequency. We first built query_bedpe to quickly access the conformation matrix and obtain interaction counts between any two regions. Over time, we’ve enhanced its abilities to handle imprecise loop coordinates.
For this primer we’re looking at two example interacting regions, each 55kb in size, separated by a distance of 120kb. These two regions are stored in a bedpe file.
bedpe=$/path/to/sample.bedpe
cat $bedpe
chr16 875000 930000 chr16 1050000 1105000Before we continue with query_bedpe, let’s first visualize this hypothetical loop using another tool in the AQuA Tools suite, plot_contacts. Visualizations will be helpful understanding the outputs we get from query_bedpe. To accomplish this, we will use the RH4_DMSO_K27ac sample at 5kb resolution.
annotation=$tutorial_data_path/sample.bed
cat $annotation
chr16 875000 930000
chr16 1050000 1105000
range=chr16:790000:1260000:test
resolution=5000
genome=hg38
plot_contacts \
-A $sample1 \
-R $range \
-G $genome \
-O AL_1.pdf \
-Q aqua \
--max_cap 1 \
--annotations_default FALSE \
--annotations_custom $annotation \
--annotations_custom_color c000ff \
--bedpe $bedpe \
--bedpe_color c000ff
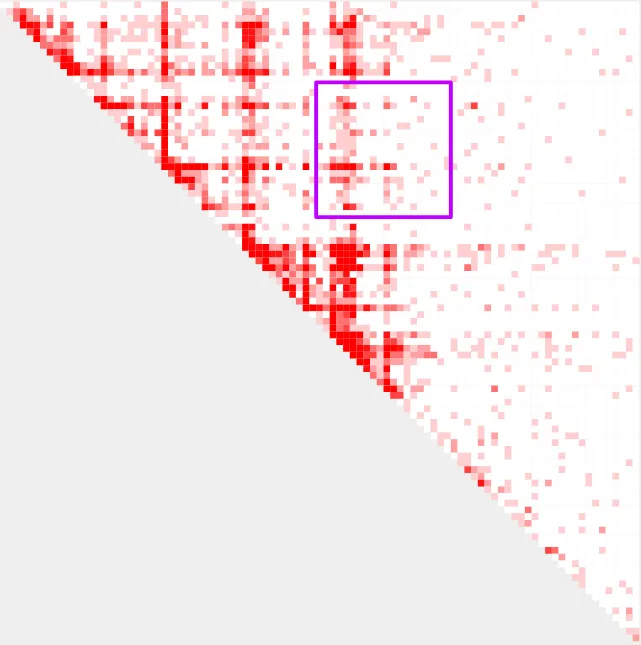
This is our loop!
Reporting bin values with —formula
Now that we have a visual understanding of the loop we are working with, we can use query_bedpe to ask questions about this loop. We are likely interested in the contact values (red bins) within this region, but which contact value from the loop (purple square) should we report? The default option is the center bin.


However, in this case, the default (center) bin is not the best choice. For single sample analyses, query_bedpe will return coordinates in the first 6 columns and contact values in the 7th column. As you can see below, query_bedpe reports a 0 value for this bin.
query_bedpe \
- A $sample1 \
- P $bedpe \
- G $genome
chr16 875000 930000 chr16 1050000 1105000 0The default can be changed to obtain the max, sum or average of the enclosed 2D area, using the -f or --formula parameter.
query_bedpe \
-A $sample1 \
-P $bedpe \
-G $genome \
--formula max
chr16 875000 930000 chr16 1050000 1105000 2.42query_bedpe \
-A $sample1 \
-P $bedpe \
-G $genome \
--formula sum
chr16 875000 930000 chr16 1050000 1105000 11.54query_bedpe \
-A $sample1 \
-P $bedpe \
-G $genome \
--formula average
chr16 875000 930000 chr16 1050000 1105000 0.095This is the area for sum and average --formula parameters
This is the area for max --formula parameter

Normalization methods
We can also change default behavior of the tool and report counts per million contact values, instead of the default AQuA normalized values, using the -Q or
--norm parameter.
query_bedpe \
-A $sample1 \
-P $bedpe \
-G $genome \
--formula max \
--norm cpm
chr16 875000 930000 chr16 1050000 1105000 0.436query_bedpe \
-A $sample1 \
-P $bedpe \
-G $genome \
--formula sum \
--norm cpm
chr16 875000 930000 chr16 1050000 1105000 2.079query_bedpe \
-A $sample1 \
-P $bedpe \
-G $genome \
--formula average \
--norm cpm
chr16 875000 930000 chr16 1050000 1105000 0.017Fixed or updating coordinates based on —formula
The above example reports back the same bedpe coordinates you supplied. However, the center and max bins (contained within the supplied 2D area) will have different (smaller) 2D genomic areas than the ones supplied. Using the -F or --fix parameter, we can instruct query_bedpe to either keep the coordinates fixed (default; —fix TRUE), or to return updated bedpe coordinates along with the contact value (—fix FALSE). This assists in obtaining precise coordinates from fuzzy intervals. Let’s compare output when --fix is TRUE and FALSE.
query_bedpe \
-A $sample1 \
-P $bedpe \
-G $genome \
--formula center \
--fix FALSE
chr16 900000 905000 chr16 1075000 1080000 0
# --fix TRUE
# chr16 875000 930000 chr16 1050000 1105000 0query_bedpe \
-A $sample1 \
-P $bedpe \
-G $genome \
--formula max \
--fix FALSE
chr16 910000 915000 chr16 1050000 1055000 0.436
# --fix TRUE
#chr16 875000 930000 chr16 1050000 1105000 0.436query_bedpe \
-A $sample1 \
-P $bedpe \
-G $genome \
--formula sum \
--fix FALSE
chr16 875000 930000 chr16 1050000 1105000 2.079
# --fix TRUE
# chr16 875000 930000 chr16 1050000 1105000 2.079query_bedpe \
-A $sample1 \
-P $bedpe \
-G $genome \
--formula average \
--fix FALSE
chr16 875000 930000 chr16 1050000 1105000 0.017
# --fix TRUE
#chr16 875000 930000 chr16 1050000 1105000 0.017You can see how the coordinates have changed when using --fix FALSE for center and max bins.
Two sample query & delta contact values
We can also use query_bedpe to obtain contact values of bedpe regions of two samples at the same time.
query_bedpe \
-A $sample1 \
-P $bedpe \
-G $genome \
-B $sample2
chr16 875000 930000 chr16 1050000 1105000 0 0.379 0.379We now have two new columns! The first contact value is for sample1 (-A), the second for sample2 (-B), and the third column is the delta contact value (sample2 - sample1). This can be very useful in case-control analyses.
query_bedpe \
-A $sample1 \
-P $bedpe \
-G $genome \
-B $sample2 \
--formula max
chr16 875000 930000 chr16 1050000 1105000 2.42 2.653 0.233What happens if we supply --fix FALSE to get the precise max bin coordinates?
query_bedpe \
- A $sample1 \
- P $bedpe \
- G $genome \
- B $sample2 \
- -formula max \
- -fix FALSE
chr16 875000 930000 chr16 1050000 1100000 2.42 2.653 0.233We do not get altered coordinates, since we’re dealing with two samples at the same time! Parameter --fix FALSE only works with one sample analysis.
Shrink-wrap
Time to try out some more powerful query_bedpe functionalities, all designed to find better signal from fuzzy intervals! Let’s look at a larger region.
bedpe=sample_bedpe_2.bedpe
cat $bedpe
chr16 895000 990000 chr16 1020000 1070000
annotation=sample_bed_2.bed
cat $annotation
chr16 895000 990000
chr16 1020000 1070000plot_contacts \
-A $sample1 \
-R $range \
-G $genome \
-O AL_2.pdf \
-Q aqua \
--max_cap 1 \
--annotations_default FALSE \
--annotations_custom $annotation \
--annotations_custom_color c000ff \
--bedpe $bedpe \
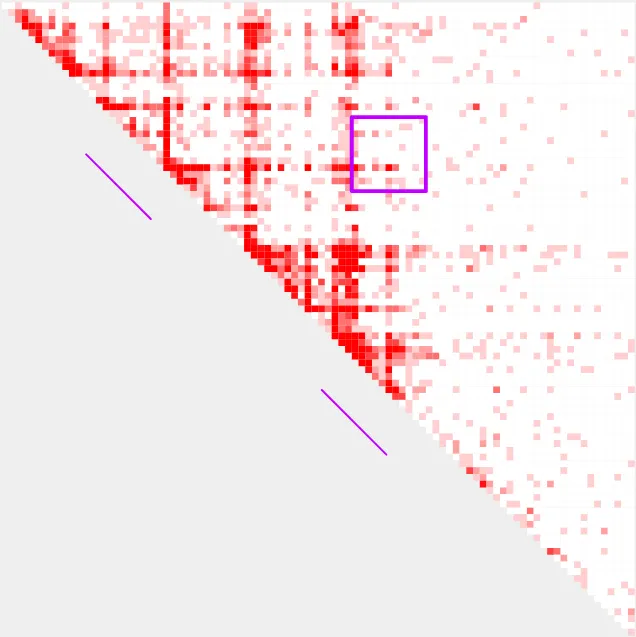
--bedpe_color c000ff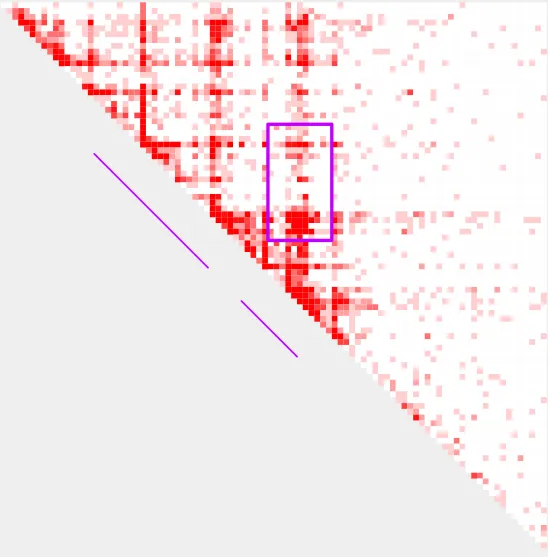
Parameter --shrink_wrap can reduce the dimensions of a 2D bedpe region until it encounters a bin with a user-supplied count value. Here, we will use 10 as the count value. We will also use
--formula sum to show the decrease following --shrink_wrap, since the 2D area is shrinking.
query_bedpe \
- A $sample1 \
- P $bedpe \
- G $genome \
- -formula sum
chr16 895000 990000 chr16 1020000 1070000 62.744
query_bedpe \
- A $sample1 \
- P $bedpe \
- G $genome \
- -formula sum \
- -shrink_wrap 10 > shrink.bedpe
cat shrink.bedpe
chr16 910000 990000 chr16 1040000 1060000 46.173Let’s visually inspect the effect of shrink_wrap with plot_contacts
plot_contacts \
-A $sample1 \
-R $range \
-G $genome \
-O AL_3.pdf \
-Q aqua \
--max_cap 2 \
--annotations_default FALSE \
--bedpe shrink.bedpe \
--bedpe_color c000ffbefore shrink_wrap

after shrink_wrap

Split
Parameter --split can identify clusters of bins above a user-supplied contact value. The size of the cluster can be controlled by the parameter --padding.
query_bedpe \
- A $sample1 \
- P $bedpe \
- G $genome \
- -split 8 --padding 2 > split.bedpe
cat split.bedpe
chr16 900000 925000 chr16 1040000 1065000 2.42
chr16 960000 1000000 chr16 1030000 1070000 3.91plot_contacts \
-A $sample1 \
-R $range \
-G $genome \
-O AL_4.pdf \
-Q aqua \
--max_cap 1 \
--annotations_default FALSE \
--bedpe split.bedpe \
--bedpe_color c000ffbefore split
after split

Expand
Given a supplied 2D bedpe area, parameter --expand can, as the name suggests, expand the 2D area in units of the resolution. We will use --expand with --fix FALSE to see the expanded coordinates in our output. Let’s use our original loop example again.
bedpe=sample_bedpe.bedpe
cat $bedpe
chr16 875000 930000 chr16 1050000 1105000
query_bedpe \
- A $sample1 \
- P $bedpe \
- G $genome \
- -formula sum
chr16 875000 930000 chr16 1050000 1105000 11.543
query_bedpe \
- A $sample1 \
- P $bedpe \
- G $genome \
- -formula sum \
- -fix FALSE \
- -expand 5
chr16 850000 955000 chr16 1025000 1130000 33.699before expand
after expand